In the past several weeks I have been using MaSuRCA to create improved assemblies of several genomes of varying difficulty and I was able to detect and fix several performance issues and bugs:
1. run time too long in scaffolder and consensus when using Illumina linking mate pairs or other mater reads such as Sanger or 454
2. failure when combining single end and paired end paired end libraries when USE_LINKING_MATES is turned on
3. accuracy and contiguity of the assembly does not improve uniformly with more PacBio data
4. overlap filtering step after the unitigger takes too long for large data sets
5. mapping in duplicate/haplotype filtering is faster, because I excluded the singleton unitigs from the mapping
6. Illumina mate pairs are now properly used in scaffolding of assemblies that contain PacBio or Nanopore reads
Thanks to all MaSuRCA users who reported their issues to me and I hope I was able to address most of them.
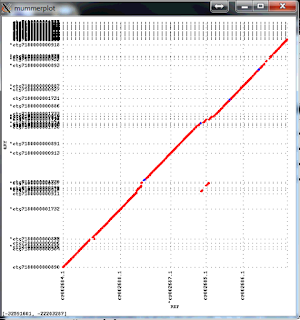 The resulting package is much faster and more accurate. Here is an example of an assembly of Arabidopsis thaliana data set with 50x PacBio data, aligned to the finished genome. Note that the finished sequence is different species but there should not be any large structural differences. The plot shows a line for each contig with start and end positions corresponding tot the alignment positions in the finished genome (X axis) and in the assembly (Y axis). There are three off-diagonal matches which correspond to three small misassemblies. The N50 contig size of this assembly is 8Mb, which is close to about 10Mb one can achieve with 100x PacBio data. The NGA50 -- N50 after breaking at misassemblies is 7.1Mbp.
The resulting package is much faster and more accurate. Here is an example of an assembly of Arabidopsis thaliana data set with 50x PacBio data, aligned to the finished genome. Note that the finished sequence is different species but there should not be any large structural differences. The plot shows a line for each contig with start and end positions corresponding tot the alignment positions in the finished genome (X axis) and in the assembly (Y axis). There are three off-diagonal matches which correspond to three small misassemblies. The N50 contig size of this assembly is 8Mb, which is close to about 10Mb one can achieve with 100x PacBio data. The NGA50 -- N50 after breaking at misassemblies is 7.1Mbp.
I also changed the way super-reads are mapped to the PacBio and Nanopore reads. The problem was in connectivity of the super-reads -- to snap two super-reads together to create a mega-read, an overlap of K or more is needed, and we try to maximize K to create longer super-reads e.g. K could be 127bp. For example we have super-reads A_B_C, C_D and D_E_F. To connect super-reads A_B_C and D_E_F to create the mega-read A_B_C_D_E_F, we need to have A_B_C and D_E_F to have exact overlaps with C_D. So if for some reason C_D is lost in the mapping a gap may be created. But the gap does not need to be there because A_B_C and D_E_F may still overlap exactly, with the overlap shorter than K. Now the super-reads are pre-processed to reduce K to 41 which effectively reduces the minimum overlap length for creating mega-reads from super-reads while still keeping the principle of the algorithm intact.
I feel that MaSuRCA mega-reads is not mature enough and tested to give it Release Candidate status. The new MaSuRCA is available here:
ftp://ftp.genome.umd.edu/pub/MaSuRCA/latest/MaSuRCA-3.2.2_RC1.tar.gz
Please post any issues with the new version in comments to this blog post.
1. run time too long in scaffolder and consensus when using Illumina linking mate pairs or other mater reads such as Sanger or 454
2. failure when combining single end and paired end paired end libraries when USE_LINKING_MATES is turned on
3. accuracy and contiguity of the assembly does not improve uniformly with more PacBio data
4. overlap filtering step after the unitigger takes too long for large data sets
5. mapping in duplicate/haplotype filtering is faster, because I excluded the singleton unitigs from the mapping
6. Illumina mate pairs are now properly used in scaffolding of assemblies that contain PacBio or Nanopore reads
Thanks to all MaSuRCA users who reported their issues to me and I hope I was able to address most of them.

I also changed the way super-reads are mapped to the PacBio and Nanopore reads. The problem was in connectivity of the super-reads -- to snap two super-reads together to create a mega-read, an overlap of K or more is needed, and we try to maximize K to create longer super-reads e.g. K could be 127bp. For example we have super-reads A_B_C, C_D and D_E_F. To connect super-reads A_B_C and D_E_F to create the mega-read A_B_C_D_E_F, we need to have A_B_C and D_E_F to have exact overlaps with C_D. So if for some reason C_D is lost in the mapping a gap may be created. But the gap does not need to be there because A_B_C and D_E_F may still overlap exactly, with the overlap shorter than K. Now the super-reads are pre-processed to reduce K to 41 which effectively reduces the minimum overlap length for creating mega-reads from super-reads while still keeping the principle of the algorithm intact.
I feel that MaSuRCA mega-reads is not mature enough and tested to give it Release Candidate status. The new MaSuRCA is available here:
ftp://ftp.genome.umd.edu/pub/MaSuRCA/latest/MaSuRCA-3.2.2_RC1.tar.gz
Please post any issues with the new version in comments to this blog post.