Dear Users, I wanted to stress one more time that there have been significant improvements in speed (>2x), accuracy and disk usage footprint (reduced by about factor of 10), as well as many bugfixes in the MaSuRCA 3.2.2 release compared to the previous 3.2.2_RCX and 3.2.1_XXXXXXXX versions. Please avoid using previously released beta-versions, now that a release is available. I urge you to take a moment to update your installation of MaSuRCA to the latest version available here: ftp://ftp.genome.umd.edu/pub/MaSuRCA/latest/MaSuRCA-3.2.2.tar.gz
Friday, May 19, 2017
Monday, May 15, 2017
Human NA12878 hybrid 30x MinION+100x Illumina assembly by MaSuRCA 3.2.2
As a final test of the release version of the MaSuRCA 3.2.2 assembler, I created an assembly of the human NA12878 data set from ~30x coverage of Oxford Nanopore data (rel3, https://github.com/nanopore-
The hybrid assembly took about 50,000 CPU-hours on my AMD Opteron 6000-series 400-core cluster. The assembly had the following quantitative statistics(G=3Gb):
Update 05/23/2017: Just for the sake of comparison here are the stats of the MaSuRCA assembly that used only the Illumina data:
Addition of the nanopore data makes huge difference in the assembly contiguity.
Compared to the available Canu Nanopore-only assembly of the rel3 data, The MaSuRCA assembly has 37% bigger N50 contig size, and has about 9% more sequence:
https://genomeinformatics.
The MaSuRCA assembly aligns to the GRCh38.p10 human reference (this is not the same human) at average 99.73% identity for 1-to-1 alignments.
The 1-to-1 alignments to Illumina-only assembly of the NA12878 have an average identity of 99.96%, implying consensus error rate of 4 errors per 10,000 bases. Below is an alignment plot created by mummerplot of the biggest scaffold in the assembly (22.4Mb) aligning to the corresponding GRCh38 chromosome, that shows excellent large scale agreement:
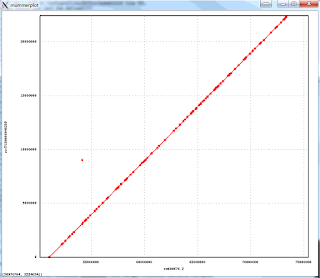
The assembly is available here:
ftp://ftp.genome.umd.edu/pub/NA12878/assembly.fa
I am now working on validation of the structure of the assembly and classification of consensus errors.
Data used. For this assembly I used the publicly available Oxford Nanopore MinION data set rel3:
https://github.com/nanopore-
and about 100x coverage by Illumina 2x150b HiSeq 2500 Paired End reads from the Genome In A Bottle project:
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/NA12878/NIST_NA12878_HG001_HiSeq_300x .
I used the following Illumina runs from that FTP site:
131219_D00360_005_BH814YADXX
131219_D00360_005_AN81VLADXX
140115_D00360_0009_AH8962ADXX
140207_D00360_0013_AH8G92ADXX
The hybrid assembly took about 50,000 CPU-hours on my AMD Opteron 6000-series 400-core cluster. The assembly had the following quantitative statistics(G=3Gb):
Sequence in scaffolds: 2.88Gb
NG50 scaffold size: 5.04Mb
NG50 contig size: 4.06Mb
Update 05/23/2017: Just for the sake of comparison here are the stats of the MaSuRCA assembly that used only the Illumina data:
Sequence in scaffolds: 2.81Gb
NG50 scaffold size: 0.065Mb
NG50 contig size: 0.068Mb
Addition of the nanopore data makes huge difference in the assembly contiguity.
Compared to the available Canu Nanopore-only assembly of the rel3 data, The MaSuRCA assembly has 37% bigger N50 contig size, and has about 9% more sequence:
https://genomeinformatics.
The MaSuRCA assembly aligns to the GRCh38.p10 human reference (this is not the same human) at average 99.73% identity for 1-to-1 alignments.
The 1-to-1 alignments to Illumina-only assembly of the NA12878 have an average identity of 99.96%, implying consensus error rate of 4 errors per 10,000 bases. Below is an alignment plot created by mummerplot of the biggest scaffold in the assembly (22.4Mb) aligning to the corresponding GRCh38 chromosome, that shows excellent large scale agreement:
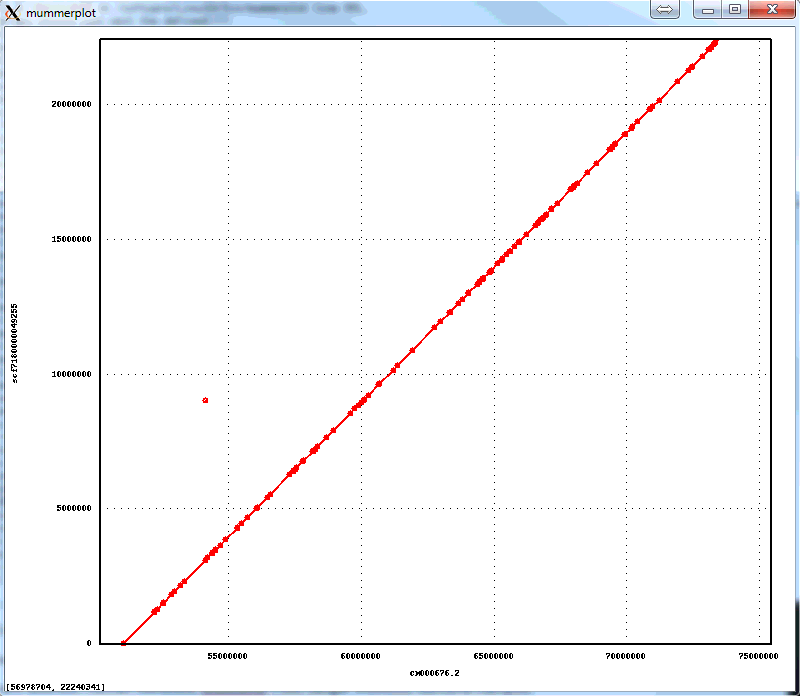
The assembly is available here:
ftp://ftp.genome.umd.edu/pub/NA12878/assembly.fa
I am now working on validation of the structure of the assembly and classification of consensus errors.
Data used. For this assembly I used the publicly available Oxford Nanopore MinION data set rel3:
https://github.com/nanopore-
and about 100x coverage by Illumina 2x150b HiSeq 2500 Paired End reads from the Genome In A Bottle project:
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/NA12878/NIST_NA12878_HG001_HiSeq_300x .
I used the following Illumina runs from that FTP site:
131219_D00360_005_BH814YADXX
131219_D00360_005_AN81VLADXX
140115_D00360_0009_AH8962ADXX
140207_D00360_0013_AH8G92ADXX
Thursday, May 4, 2017
New version MaSuRCA release 3.2.2
Today I posted a new version that I can finally call a "release" version 3.2.2. You can get it here ftp://ftp.genome.umd.edu/pub/MaSuRCA/latest/MaSuRCA-3.2.2.tar.gz . This version has numerous speed and stability improvements based on feedback from the users and my own experience running assembly experiments.
Here are the major changes, in addition to many bugfixes based on the user feedback:
1. based on the user feedback, disabled stones and Extend Clear Ranges steps in CABOG, because they have been failing and/or causing long run times without significant benefit on assemblies with long read PacBio/MinION data.
2. introduced speedups in creation of the mega-reads a long read that yields a single mega-read in pass 1 is not re-processed in pass 2. This reduced the mega-read correction time by about 25%.
3. introduced contained reads filter before CABOG; by nature of the mega-reads creation from PacBio/MinION reads, many mega-reads end up being exactly contained in other mega-reads and thus they do not contribute any new information in the assembly. I found an efficient way to remove these exact containees, reducing the coverage and the number of reads that CABOG has to deal with.
This improved the run time of CABOG by about factor of 4. Now 120Mb plant genome with 30x PacBio coverage and 100x Illumina coverage assembles in about 8 hours on a single 48-core AMD Opteron server; 12Mb yeast data with about the same level of coverage assembles in under 1 hour on the same server.
4. reworked the way assembly failures are reported making the error messages more informative
5. fixed bugs in SOAPDenovo2 assembly module thanks to the input from Rubang Luo, author of SOAPDenovo2.
6. removed dependency on "parallel" that has been causing problems for many users
Thanks to all users who reported their failures and successes to me. Your feedback is extremely valuable, it helps me make MaSuRCA more stable and easier to use!
Here are the major changes, in addition to many bugfixes based on the user feedback:
1. based on the user feedback, disabled stones and Extend Clear Ranges steps in CABOG, because they have been failing and/or causing long run times without significant benefit on assemblies with long read PacBio/MinION data.
2. introduced speedups in creation of the mega-reads a long read that yields a single mega-read in pass 1 is not re-processed in pass 2. This reduced the mega-read correction time by about 25%.
3. introduced contained reads filter before CABOG; by nature of the mega-reads creation from PacBio/MinION reads, many mega-reads end up being exactly contained in other mega-reads and thus they do not contribute any new information in the assembly. I found an efficient way to remove these exact containees, reducing the coverage and the number of reads that CABOG has to deal with.
This improved the run time of CABOG by about factor of 4. Now 120Mb plant genome with 30x PacBio coverage and 100x Illumina coverage assembles in about 8 hours on a single 48-core AMD Opteron server; 12Mb yeast data with about the same level of coverage assembles in under 1 hour on the same server.
4. reworked the way assembly failures are reported making the error messages more informative
5. fixed bugs in SOAPDenovo2 assembly module thanks to the input from Rubang Luo, author of SOAPDenovo2.
6. removed dependency on "parallel" that has been causing problems for many users
Thanks to all users who reported their failures and successes to me. Your feedback is extremely valuable, it helps me make MaSuRCA more stable and easier to use!
Subscribe to:
Posts (Atom)